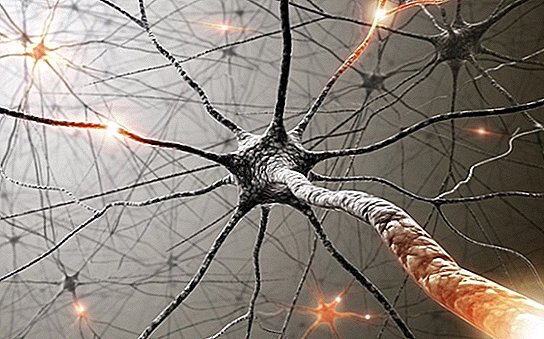
Amyotrophe Lateralsklerose (ALS), auch bekannt als Lou-Gehrig-Krankheit oder Motoneuron-Krankheit, ist eine fortschreitende neurologische Erkrankung, die nach Angaben der National Institutes of Health (NIH) zu einer Degeneration der Neuronen führt, die die freiwilligen Muskeln (Motoneuronen) steuern.
Der Begriff "Lou Gehrig-Krankheit" ist nach dem berühmten amerikanischen Baseballspieler benannt, der 1939 im Alter von 36 Jahren an ALS erkrankte. In den Vereinigten Staaten leiden 20.000 bis 30.000 Menschen an der Krankheit, und jedes Jahr wird bei etwa 5.000 Menschen eine Krankheit diagnostiziert nach Angaben der Zentren für die Kontrolle und Prävention von Krankheiten (CDC).
ALS trifft häufig Menschen im Alter von 40 bis 60 Jahren. Es betrifft Menschen aller Rassen und Ethnien. Die Krankheit ist bei Männern etwas häufiger als bei Frauen, aber der Unterschied nimmt ab.
Die Behandlungen für die Krankheit sind begrenzt, aber vielversprechende Forschung ist im Gange.
ALS-Symptome
Die ersten Symptome sind normalerweise Schwäche oder verspannte und steife Muskeln (Spastik) in einem bestimmten Bereich, sagte Dr. Jaydeep Bhatt, Neurologe am NYU Langone Medical Center in New York City. Andere Symptome sind verschwommene und nasale Sprache sowie Schwierigkeiten beim Kauen oder Schlucken.
Wenn ALS in den Armen oder Beinen beginnt, wird es als "Gliedmaßenbeginn" ALS bezeichnet. Jemand mit der Krankheit hat möglicherweise Probleme beim Schreiben oder Knöpfen eines Hemdes oder hat das Gefühl, beim Gehen oder Laufen zu stolpern oder zu stolpern. Bei Patienten, bei denen die Sprache zuerst betroffen ist, wird die Krankheit als "Bulbar Onset" ALS bezeichnet.
Mit fortschreitender Krankheit breitet sich Schwäche oder Atrophie im ganzen Körper aus. Patienten können Probleme haben, sich zu bewegen, zu schlucken und zu sprechen. Eine ALS-Diagnose erfordert Anzeichen einer Schädigung des oberen und unteren Motoneurons. Anzeichen des ersteren sind Muskelverspannungen oder -steifheit und abnormale Reflexe; Anzeichen für Letzteres sind Muskelschwäche, Krämpfe, Zuckungen und Atrophie.
Schließlich verlieren Menschen mit ALS die Fähigkeit zu stehen oder zu gehen, ihre Hände und Arme zu benutzen oder normal zu essen. In späten Stadien der Krankheit erschwert eine Schwäche der Atemmuskulatur das Atmen ohne Beatmungsgerät. Die kognitiven Fähigkeiten bleiben größtenteils intakt, obwohl einige Personen Probleme mit dem Gedächtnis oder der Entscheidungsfindung haben oder Anzeichen von Demenz zeigen können.
Lebenserwartung
Die meisten Menschen mit ALS sterben innerhalb von drei bis fünf Jahren nach Auftreten der Symptome an Atemstillstand, obwohl laut NIH etwa 10 Prozent der Betroffenen 10 oder mehr Jahre leben.
"Es gibt viele Cousins von ALS, die milder sein können", sagte Bhatt. "Manchmal ist es für einen Arzt schwierig zu unterscheiden, welches welches ist. Wir haben keinen Blut- oder MRT-Test", fügte er hinzu.
Der theoretische Physiker und Kosmologe Stephen Hawking, der an einer Motoneuronerkrankung im Zusammenhang mit ALS leidet, hat seit seiner Diagnose im Alter von 21 Jahren trotz einer anfänglichen Lebenserwartung von nur wenigen Jahren mehr als 50 Jahre überlebt.
Ursachen von ALS
Die Ursache von ALS ist unbekannt, obwohl einige Fälle - in denen eine Familiengeschichte der Krankheit vorliegt - mit Mutationen im Gen für ein Enzym namens SOD1 verbunden sind. Es ist nicht klar, wie die Mutationen die Degeneration von Motoneuronen verursachen, aber Studien legen nahe, dass das SOD1-Protein toxisch werden kann.
Laut einer Studie von HC Miranda und AR La Spada aus dem Jahr 2016, Forschern der Universität von Kalifornien in San Diego, wird angenommen, dass ungefähr 5 Prozent der ALS-Fälle genetisch bedingt sind (bekannt als familiäres ALS oder FALS), und die restlichen 95 Prozent sind sporadisch (SALS) ).
Wissenschaftler haben mehr als ein Dutzend anderer genetischer Mutationen identifiziert, die möglicherweise mit ALS zusammenhängen. Diese Mutationen verursachen Veränderungen in der Verarbeitung von RNA-Molekülen (die Gene regulieren können), Defekte beim Recycling von Proteinen, Defekte in Form und Struktur von Motoneuronen oder Anfälligkeit für Umweltgifte.
Andere Untersuchungen legen nahe, dass ALS Ähnlichkeiten mit der frontotemporalen Demenz (FTD) aufweist, einer degenerativen Erkrankung des Frontallappens des Gehirns. Ein Defekt im C9orf72-Gen wird bei einer beträchtlichen Anzahl von ALS-Patienten sowie einigen FTD-Patienten gefunden.
Eine Veröffentlichung von R. L. McLaughlin et al. Aus dem Jahr 2017 legt nahe, dass möglicherweise auch eine genetische Beziehung zwischen ALS und Schizophrenie besteht. Das betroffene Gen ist das gleiche wie das Gen, bei dem in der FTD-Studie ein Defekt festgestellt wurde.
Aaron Glatt, Chef für Infektionskrankheiten am South Nassau Communities Hospital, sagte, dass ALS und andere neurologische Störungen wie Demenz und Schizophrenie zwar genetisch ähnlich sein könnten, die Krankheiten jedoch verschiedene Bereiche des Gehirns betreffen und eine andere nicht verursachen würde. Die Association for Frontotemporal Degeneration sagt, dass etwa 30 Prozent der ALS-Patienten schließlich Anzeichen eines Frontallappenabfalls zeigen, der ähnlich wie bei FTD sein kann. Und obwohl FTD keine Auswirkungen auf die Teile des Gehirns und des Nervensystems hat, die die Körperbewegung steuern, entwickeln etwa 10 bis 15 Prozent der FTD-Patienten ALS oder ALS-ähnliche Symptome, und die Forscher sind sich immer noch nicht sicher, warum. Und umgekehrt können Menschen mit ALS möglicherweise auch einen kognitiven Rückgang ähnlich wie bei FTD erleben. Glatt sagte jedoch, dass diese Symptome typischerweise von einem Sauerstoffmangel herrühren, der das Gehirn erreicht, anstatt tatsächlich FTD zu entwickeln.
Behandlung für ALS
Derzeit hat ALS keine Heilung, aber es gibt Behandlungen, um die Symptome zu lindern und die Lebensqualität der Patienten zu verbessern.
Das erste Medikament zur Behandlung der Krankheit, Riluzol, wurde 1995 von der Food and Drug Administration (FDA) zugelassen. Es wird angenommen, dass Riluzol die Schädigung der Motoneuronen durch Minimierung der Freisetzung des chemischen Signals Glutamat verringert. In klinischen Studien verlängerte das Medikament das Überleben von ALS-Patienten (insbesondere von Patienten mit Schluckbeschwerden) um mehrere Monate. Dies kann auch die Zeit verlängern, bevor ein Patient beatmet werden muss.
Die FDA hat im Mai 2017 ein neues Medikament namens Radicava zugelassen. Es hat sich gezeigt, dass das neue Medikament die Rate, mit der Menschen mit ALS im Vergleich zu einem Placebo einen physischen Rückgang erfahren, signifikant senkt. Die verlangsamte Abnahmerate variiert nach Angaben der ALS Association in Abhängigkeit von der Fortschreitungsrate der Krankheit und der individuellen körperlichen Funktion der Patienten zu Beginn der Behandlung. Radicava wurde entwickelt, um Zellschäden zu verhindern, indem es dem Körper hilft, überschüssige freie Radikale zu eliminieren.
Ein weiteres Medikament, Nuedexta, wurde 2010 von der FDA zur Behandlung von unfreiwilligem Weinen oder Lachen zugelassen und heißt Pseudobulbar Affect. Bei ALS tritt dies auf, wenn Nerven die Gesichtsmuskeln nicht mehr kontrollieren können, was zu "emotionaler Inkontinenz" führt.
"Es ist behandelbar und jetzt gibt es ein Medikament dafür", sagte Bhatt.
Ärzte können Medikamente zur Verringerung von Müdigkeit, Muskelkrämpfen, Muskelspastik und übermäßigem Speichel oder Schleim sowie Schmerzen, Depressionen, Schlafstörungen oder Verstopfung verschreiben.
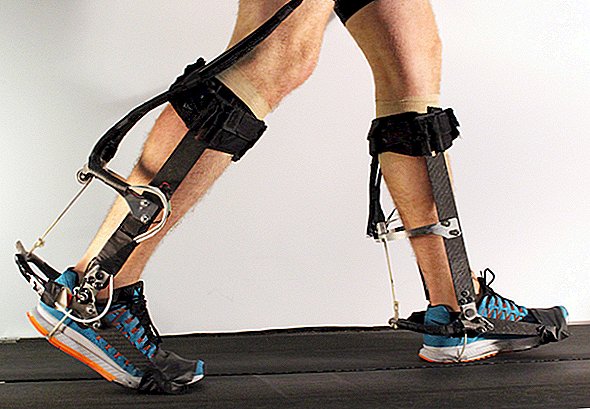
ALS beeinflusst die Atemmuskulatur, insbesondere das Zwerchfell. Ein minimalinvasives Gerät namens Zwerchfellschrittmacher, das das Zwerchfell elektrisch stimuliert, kann den Patienten beim Atmen helfen. Das Gerät kann die Lebensqualität eines Patienten verbessern, bevor ein Beatmungsgerät eingeführt wird, sagte Bhatt.
Körperliche Bewegung oder Therapie können den Patienten Unabhängigkeit geben. Zum Beispiel können Gehen, Schwimmen und stationäres Radfahren die Muskeln stärken, die nicht von der Krankheit betroffen sind, was zu einer verbesserten Herzgesundheit und weniger Müdigkeit und Depressionen führt. Spezielle Geräte wie Rampen, Zahnspangen, Gehhilfen und Rollstühle können den Patienten Mobilität bieten, ohne sie zu erschöpfen.
Logopäden und Ernährungswissenschaftler können ALS-Patienten helfen, die Probleme beim Sprechen oder Schlucken haben. Mit fortschreitender Krankheit können Patienten lernen, Ja-oder-Nein-Fragen mit den Augen zu beantworten.
Wenn das Atmen schwierig wird, können Personen mit ALS Beatmungsgeräte verwenden, die ihre Lunge während der Nacht oder schließlich ganztägig künstlich aufblasen. Atemschutzgeräte, die direkt an die Luftröhre angeschlossen sind, können letztendlich verwendet werden.
Klinische Forschung
In den letzten Jahren wurden Fortschritte bei der Entwicklung unterstützender Technologien erzielt, einschließlich Schnittstellen zwischen Gehirn und Computer. Diese Geräte zeichnen die elektrischen Signale des Gehirns auf und übersetzen sie in Befehle, mit denen Computercursor oder Prothesen gesteuert werden können. Diese Systeme sind jedoch noch nicht für den klinischen Einsatz verfügbar.
Ein weiteres vielversprechendes Forschungsgebiet war die Untersuchung der Verwendung von Stammzellen, Zellen, die das Potenzial haben, sich zu jedem Gewebetyp, einschließlich Hirngewebe, zu entwickeln. Stammzellen können in einem Labor zu Neuronen gezüchtet werden, aber es bleibt eine Herausforderung, sie sicher und effektiv in einem Menschen wachsen zu lassen, sagte Bhatt.
Die Stammzellenforschung ist laut Glatt äußerst vielversprechend, wird aber erst in einigen Jahren verfügbar sein. Ein Artikel von P. Petrou et al. Aus dem Jahr 2016 hat gezeigt, dass eine neue Stammzellenbehandlung mit eigenen Stammzellen aus dem Knochenmark das Fortschreiten der Krankheit bei einer Gruppe von 26 Patienten verlangsamt hat. Forscher des Harvard Stem Cell Institute konnten erfolgreich Stammzellen herstellen, um neue Therapien aus Haut und Blut von ALS-Patienten zu untersuchen.
Die ALS Association sagt, dass Stammzellen möglicherweise Wachstumsfaktoren oder Schutz für vorhandene Motoneuronen im Rückenmark bieten. Eines Tages können jedoch Stammzellen verwendet werden, um die sterbenden Motoneuronen zu ersetzen, die die Herausforderung überwinden, die Neuronen in geeigneter Weise mit den umgebenden Muskeln zu verbinden.
Leider jagen einige Ärzte Patienten, indem sie unwirksame Stammzellbehandlungen verkaufen, sagte er. Die Realität ist: "Es ist sehr schwierig, leicht wachsende motorische Nerven in einem Reagenzglas in eine Person zu bringen."
Das Bewusstsein für ALS nimmt zu. An der "ALS Ice Bucket Challenge" 2014 nahmen Einzelpersonen Eiswasser auf den Kopf, um das Bewusstsein zu fördern. Die Kampagne wurde viral und bis Dezember 2014 hatte die ALS Association Spenden in Höhe von 115 Millionen US-Dollar gesammelt.